Thermodynamics-Part1
Overview
Thermodynamics is a branch of physics which deals with the energy and work of a system. Thermodynamics deals only with the large scale response of a system which we can observe and measure in experiments.
Work
In defining work, we focus on the effects that the system has on its surroundings. Thus we define work as positive when the system does work on the surroundings, vice versa.
Consider a simple compressible substance, a gas (the system) exerting a force on the surrounding via a piston, which moves through some distance, l. The work done on the surroundings: dW = px * dV.
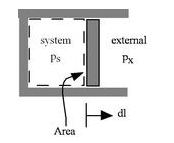
Why is the pressure px instead of ps? From the definition of work, work is the energy it takes to move an object against a resistant force. Consider the case px = 0. Since there is no resistance, no work is done on the surroundings even though ps changes and the system volume changes. Thus, it is px, the external pressure that directly related to the work.
However, use of px instead of ps is often incovenient because it is usually the state of the system that we are interested in. The external pressure can only be related to the system pressure if px is close to ps. For this to occur, there cannot be any friction, and the process must also be slow enough so that pressure differences due to accelerations are not significant. In other words, we require a reversible process (quasi-static + frictionless) so that ps is almost the same as px. And the 'work done by the system' is the same as the 'work done on the sourroundings'. Under these conditions, we say that the process is 'reversible'. In the case of a 'free expansion', where px = 0, ps is not related to px and thus not related to the work because the system is not in equilibrium.
For a reversible process, we can substitute dW = ps*dV. For irreversible processes, we cannot use dW = ps*dV. Either the work must be given or it must be found by another method.
Zeroth law of Thermodynamics
The zeroth law of thermodynamics begins with a simple definition of thermodynamic equilibrium . It is observed that some property of an object, like the pressure in a volume of gas, the length of a metal rod, or the electrical conductivity of a wire, can change when the object is heated or cooled. If two of these objects are brought into physical contact there is initially a change in the property of both objects. But, eventually, the change in property stops and the objects are said to be in thermal, or thermodynamic, equilibrium. Thermodynamic equilibrium leads to the large scale definition of temperature. When two objects are in thermal equilibrium they are said to have the same temperature.
Equation of State
Gases have various properties that we can observe with our senses, including the gas pressure p, temperature T, mass m, and volume V. Careful, scientific observation has determined that these variables are related to one another, and the values of these properties determine the state of the gas. If we fix any two of the properties we can determine the nature of the relationship between the other two.
The equation of state can be written in terms of the specific volume as: p * v = R * T. Notice that the equation of state given here applies only to an ideal gas, or a real gas that behaves like an ideal gas. There are in fact many different forms for the equation of state for different gases.
First Law of Thermodynamics
There are three principal laws of thermodynamics. Each law leads to the definition of thermodynamic properties which help us to understand and predict the operation of a physical system.
There exists for every system a property called energy, E. The system energy can be considered as a sum of internal energy, kinetic energy, potential energy, and chemical energy. he First Law describes that: The change in energy of a system (delta E) is equal to the difference of the heat transfer (Q) into a system and the work (W) done by the system, delta E = Q - W.
In many situations the potential energy, kinetic energy, and chemical energy of the system are constant or not important. Then delta E = detla U and delta U = Q - W.
Internal energy arises from the random or disorganized motion of molecules in the system. Since this molecular motion is primarily a function of temperature, it is sometimes called 'thermal energy'. The internal energy is just a form of energy like the potential energy of an object at some height above the earth, or the kinetic energy of an object in motion. In the same way that potential energy can be converted to kinetic energy while conserving the total energy of the system, the internal energy of a thermodynamic system can be converted to either kinetic or potential energy.
The first law of thermodynamics allows for many possible states of a system to exist, but only certain states are found to exist in nature. The second law of thermodynamics helps to explain this observation.
Specific Heat
In general, the amount of heat transfer is related to Temperature difference by: Q = C * delta T, where C is a constant called heat capacity and depends on the substance. Since heat is path dependant, heat capacity (C) also depends on the process. Thus, heat capacity is not a state variable.
On the other hand, Cp and Cv, which are derived from the constant pressure and constant volume processes, are state variables. Let's consider the derivation of Cv. Remember that if we specify any two properties of the system, then the state of the system is fully specified. In other words we can write internal energy u as: u = u(T,v), u = u(p, v) or u = u(p, T). Here, we use the form of u = u(T,v). Use the chain rule to write how u changes with respect to T and v: du = (delta u / delta T |v) * dT + (delta u / delta v |T) * dv. Write the First Law considering only internal energy: du = dq - dW. Assume quasi-state process (reversible), the equation becomes: du = dq - p*dv. For a costant volume process, dv = 0. Thus we eventually get: dq = (delta u / delta T |v) * dT. And cv is defined as the term: (delta u / delta T |v) from the definition of heat capacity. Similarly, cp = (delta h / delat T |p).
In the derivation of cv, we considered only a constant volume process. However, it is more useful to think of cv as a certain partial derivative, which is a state variable (thermodynamic property), rather than as a quantity related to heat transfer in a special process. In fact, the derivatives above are defined at any point in any quasi-static process whether that process is constant volume, constant pressure, or neither. The names 'specific heat at constant volume' and 'specific heat at constant pressure' are therefore unfortunate misnomers; cv and cp are state variables of a substance, and by definition depend only on the state.
Specific Heat of Ideal Gas
One of the important features of an ideal gas is that its internal energy depends only upon its temperature: u = u(T), du = (delta u / delta T |v) * dT. Therefore, for ideal gas: du = cv(T) * dT. Similarly, dh = cp(T) * dT.
If cv or cp are nearly constant between two states, it is useful to simplify the calculations to: u2 - u1 = cv*(T2 - T1), h2 - h1 = Cp*(T2-T1). Otherwise, we need to account the variation of cv or cp with repect of Temperature, and use the integral form: u2 - u1 = integral( cv(T) * dT), h2 - h1 = integral( cp(T) * dT), to calculate the difference of internal energy and enthalpy between two states.
Furthermore, we can relate cp and cv of ideal gas by the equation of state with: cp - cv = R. And we define the ratio of specific heat as: gamma = cp/cv.
Enthalpy
Let's consider the first law of thermodynamics in a special case of a constant pressure process, where the work done by the gas W = p*(V2-V1); and consider only internal energy, where delta E = delta U. Thus [U2 - U1 = Q - W] will become (U2 + p*V2) - (U1 + p*V1) = Q.
Because internal energy (U), pressure (p) and volume (V) are all state variables, we are free to define additional state variables which are combinations of existing state variables. For a gas, a useful additional state variable is the enthalpy H = U + p*V. Thus, The equation above can be written as H2 - H1 = Q.
When we evaluate the energy of an object of volume v, we have to remember that the object had to push the surroundings out of the way to make room for itself. With pressure p on the object, the work required to make a place for itself is pV. This work may not be negligible and thus the total energy of a body is its internal energy plus the extra energy by having a volume V at pressure p. We call this total energy the enthalpy, H.
From our definition of heat transfer, we can represent Q by some heat capacitiy coefficient Cp times the temperatur T. So finally we have: h2 - h1 = Cp*(T2 - T1). This final equation is used to determine values of specific enthalpy for a given temperature.
Conservation of Energy
From the First Law: E2 - E1 = Q - W. Because we are considering a moving gas, the energy of the system (E) are the sum of the internal energy (U) and kinetic energy (K), and the first law equation becomes: u2 - u1 + u2 - u1 = q - w, (in the specific form, divided by math).
There are two parts to the specific work for a moving gas. Some of the work, called the shaft work (wsh) is used to move the fluid or turn a shaft, while the rest of the work goes into changing the state of the gas. For a pressure p and specific volume v, the work is given by: w = (p * v)2 - (p * v)1 + wsh. Substituting: u2 - u1 + k2 - k1 = q - (p * v)2 + (p * v)1 - wsh.
A useful additional state variable for a gas is the specific enthalpy h which is equal to: h = u + (p * v) and total specific enthalpy: ht = h + 0.5*v^2. The final, most useful, form of the energy equation is: ht2 - ht1 = q - wsh. For a compressor or power turbine, there is no external heat flow into the gas and the "q" term is set equal to zero. In the burner, no work is performed and the "wsh" term is set to zero.
Second law of Thermodynamics
We can imagine thermodynamic processes which conserve energy but which never occur in nature. For example, if we bring a hot object into contact with a cold object, we observe that the hot object cools down and the cold object heats up until an equilibrium is reached. The transfer of heat goes from the hot object to the cold object. We can imagine a system, however, in which the heat is instead transferred from the cold object to the hot object, and such a system does not violate the first law of thermodynamics. The cold object gets colder and the hot object gets hotter, but energy is conserved. Obviously we don't encounter such a system in nature and to explain this and similar observations, thermodynamicists proposed a second law of thermodynamics. Clasius, Kelvin, and Carnot proposed various forms of the second law to describe the particular physics problem that each was studying. The description of the second law stated on this slide was taken from Halliday and Resnick's textbook, "Physics". It begins with the definition of a new state variable called entropy. Entropy has a variety of physical interpretations, including the statistical disorder of the system, but for our purposes, let us consider entropy to be just another property of the system, like enthalpy or temperature.
The second law first states that there exists a useful state variable called entropy S. The change in entropy (delta S) is equal to the heat transfer delta Q divided by the temperature T, delta S = delta Q / T. In the later discussion, we will have a more accurate definition of entropy that, the change of entropy is 'only' equal to delta Q / T for 'reversible processes': dS = dQ_rev/ T.
In addition, for a given physical process, the combined entropy of the system and the environment remains a constant if the process can be reversed. Sf = Si or delta S = 0 (reversible process). An example of a reversible process is ideally forcing a flow through a constricted pipe. Ideal means no boundary layer losses. As the flow moves through the constriction, the pressure, temperature and velocity change, but these variables return to their original values downstream of the constriction. The state of the gas returns to its original conditions and the change of entropy of the system is zero. Engineers call such a process an isentropic process. Isentropic means constant entropy.
The second law states that if the physical process is irreversible, the combined entropy of the system and the environment must increase. The final entropy must be greater than the initial entropy for an irreversible process: Sf > Si or delta S > 0 (irreversible process). An example of an irreversible process is the problem discussed in the beginning. A hot object is put in contact with a cold object. The heat will transfer from high temperature to low temperature, based on the second law that delta S = -delta Q/ T_high + delta Q/ T_low > 0. Eventually they both achieve the same equilibrium temperature. If we then separate the objects they remain at the equilibrium temperature and do not naturally return to their original temperatures. The process of bringing them to the same temperature is irreversible.
Alternative way of writing first law
Let's write the first law considering only internal energy in differential form: du = dq - dw. Assume quasi-static process so that w = p*dv, we get: du = dq - p * dv. From the definition of enthalpy that h = u + p * v and dh = du + p * dv + v * dp, we eventually have: dq = dh - v * dp. This is an alterative way to write the first law of thermodynamics.
Entropy of ideal Gas
During a thermodynamic process, the temperature T of an object changes as heat Q is applied or extracted. Thus a more correct definition of the entropy S is the differential form that accounts for this variaion: dS = dQ/T. For gases, there are two possible ways to evaluate the change in entropy. In other words, if we can represent the term dQ by the state variables, we can then integrate it bewteen two states because the result will not change by the processes it takes. Notice that the term dQ
We begin by considering the constant volume, quasi-static process. Rearranging the first law to dq = du + dw. Since the first term, du = Cv * dT and the second term, dw = p * dv, we have dq = Cv * dT + p * dV. Using the ideal equation of state pv=RT, we can write p as RT/v. Eventually we have the differential equation: dq = Cv * dT + RT * dv/v. Subsitute this equation to the definition of the entropy, we get ds = Cv * dT/T + R* dv/v. Integrate this equation from state 1 to state 2, and we have: s2 - s1 = Cv* ln(T2/T1) + R*ln(v2/v1).
Another way is by considering the constant pressure, quasi-static process. Using the alternative form of first lay,dq = dh - v * dp. Since dh = Cp * dT and v can be writen as RT/p from the ideal equation of state, we get: dq = Cp * dT - RT/p * dp. Subsitute this equation to the definition of the entropy, we get ds = Cp * dT/T - R * dP/P. Integrate this equation from state 1 to state 2, and we have: s2 - s1 = Cp * ln(T2/T1) - R* ln(P2/P1). We can now determine the change in entropy for a gas!
Carnot Cycle
Carnot Cycle is an ideal cycle which generates work by absorbing and releasing heat. Since it is ideal, the entropy, after a cycle, would not change. Carnot Cycle has 4 states, let's say a, b, c, and d.
From a to b, we reduce the external pressure, while remaining the temperature constant at T1. From the First Law delta U = Q - W. We know that there must be heat (Q1) adding to the system, because the system is generating work (W>0) with delta U = 0 (because the temperature is constant). This heat transfer can be done by contacting the system with a heat reservoir at temperature T1.
From b to c, we further reduce the external pressre, but this time keeping the system isolated so that it undergoes an adiabatic process. The system is still doing work, but without additional heat input to the system. The internal energy of the system reduces by: delta U = -W. And the temperature of the gas reduce to T2.
From c to d, we increase the external pressure, while remaining the temperature constant at T2. This time, since we are doing work on the system with detla U = 0 (because temperature remains constant), we know that there must be heat (Q2) releasing from the system. This can also be done by contacting the system with another heat reservoir at temperature T2.
From d to a, we further increase the external pressure in adiabatic process. Because no additional heat is released and we are doing work on the system, the temperature of the system increases to T1. And the cycle is done.
First we can see that the net work output around a cycle is Q1 - Q2, or the shaded area in the P-V diagram. Secondly, what is the change of entropy around a cycle? We can use the equation: delta S = Q (rev)/ T. The only processes that heat transfer involve are [a to b] and [c to d]. Heat transfer in [a to b] is: Q1 = Work done from [a to b] = integral(p)*dV. Assuming an ideal gas, we can write p = RT/V. Thus Q1 = W [a to b] = R*T1*ln(Vb/Va). Similarly Q2 = W c to d = R*T2*ln(Vd/Vc). Eventually, delta S = R*T1*ln(Vb/Va) / T1 + R*T2*ln(Vd/Vc) / T2 = R* ln( Vb/Va * Vd/Vc). From some deriavation, we can see that the term Vb/Va * Vd/Vc = 1. Thus, delta S = ln(1) = 0. So the entropy around a cycle does not change!